L’expérience 2D HSQC 1H-15N (Bodenhausen et Ruben, 1980) permet de corréler
chaque azote avec le ou les hydrogènes qui lui sont liés
covalemment. Dans une protéine, chaque résidu est caractérisé
par la liaison N–H de sa fonction amide, à l’exception des
prolines (dont l’azote ne porte aucun hydrogène) et du
résidu N-terminal (l’hydrogène de la fonction amine
étant labile). Chacune des liaisons amides N–H d’une
protéine enrichie en 15N donne lieu à un pic de
corrélation sur le spectre HSQC 1H-15N.
La position du
pic dans le spectre est décrite par les déplacements chimiques du
proton et de l’azote. Le déplacement chimique traduit le « blindage »
d’un noyau par les électrons, s’opposant à
l’établissement du champ magnétique. Il dépend de
tous les facteurs qui peuvent modifier la structure ou la
géométrie des orbitales moléculaires. Plusieurs interactions physiques interviennent dont les
contributions ne sont pas encore toutes comprises ! L’interprétation
du déplacement chimique reste donc essentiellement qualitative. On peut
généralement distinguer (au moins pour la modélisation,
cf. (Oldfield,
1995))
· Les
effets à courte portée : longueurs et angles de liaison (nature
chimique du noyau), angles de torsion (d'ou l'interprétation des
déplacements chimiques en termes de structure secondaire), liaisons
hydrogène fortes...
· Les
effets à longue portée : contributions électrostatique
(dipôles voisins) et magnétique (courant de cycle aromatique,
anisotropie du carbonyle...). Ces interactions sont particulièrement
sensibles à la structure tertiaire de la protéine et cette
sensibilité est plus forte pour le proton que pour les autres noyaux.
Le spectre HSQC 1H–15N constitue une
empreinte de la protéine d’étude et de son état de repliement,
d’où son utilisation fréquente dans les études de
faisabilité en RMN (Yee et al., 2002).
Le travail présenté ici sur le domaine C-terminal de la
protéine TolA donne un aperçu des différentes situations
rencontrées, récapitulées ci-dessous. Le lecteur de la
version électronique de ce document est encouragé à
fractionner sa fenêtre de travail afin de comparer de façon
interactive les différents spectres. Le lecteur de la version
imprimée pourra se référer aux sections
mentionnées.
|
|
Situation
|
Spectre
HSQC 1H-15N
|
Référence
|
|
1
|
Domaine protéique TolAIII3 replié
|
|
Section III.A.3.
|
|
2
|
Domaine protéique TolAIII2 dénaturé (8 M
urée)
|

|
Section VII.A.
|
|
3
|
Domaine protéique TolAIII2 présentant une
hétérogénéité de conformations
(excédent de pics de corrélation)
|

|
Section III.A.2.
|
|
4
|
Domaine protéique TolAIII1 comportant une trentaine de
résidus flexibles (raies larges au centre du spectre)
|

|
Section III.A.1.
|
|
5
|
Domaine TolAIII1 se liant à TolB (disparition de certains pics
de corrélation)
|

|
Section IV.B.1.
|
|
6
|
Domaine protéique TolAIII3 perturbé par la liaison avec
g3p N1
|

|
Section IV.A.1.
|
|
7
|
Domaine protéique TolAIII3 rendu plus flexible par la liaison
avec ATh
|

|
Section
IV.A.1.
|
|
8
|
Domaine protéique TolAIII3 rendu plus flexible par la liaison
avec ATh (57 °C)
|

|
Section
IV.A.2.
|
Tableau 8 : Galerie
de spectres HSQC 1H-15N de TolAIII à 27 °C
(sauf 8)

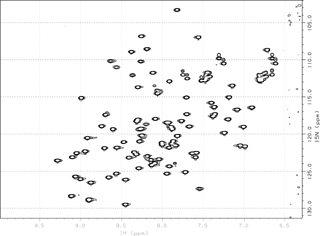
Figure 32 : Spectre HSQC 1H–15N de TolAIII2 (0,2 mM) dénaturé dans
l’urée (8 M).
L’attribution séquentielle des résonances
1H et 15N de la chaîne principale de la protéine TolAIII2 a été
réalisée à partir des expériences 3D TOCSY-HSQC 1H–15N
et 3D NOESY-HSQC 1H–15N (Marion et al., 1989),
(Bodenhausen et Ruben, 1980).
L’ajout d'une troisième dimension 1H
à un spectre HSQC
1H–15N (figure
33) permet de corréler chaque proton amide avec les protons qui
appartiennent au même résidu (3D TOCSY-HSQC 1H–15N)
ou avec les protons les plus proches dans l'espace (3D NOESY-HSQC 1H–15N).

Figure 33 : Représentation
schématique d’un spectre 3D TOCSY-HSQC 1H–15N
et de l’extraction des bandes correspondant aux systèmes de spin.
Un spectre tridimensionnel étant difficilement
exploitable directement, nous en avons extrait pour chaque N–H peptidique
repéré dans le spectre HSQC une “bande” dans la
troisième dimension. La série de bandes ainsi extraite a
été imprimée pour l’analyse
(figure 33).
Le spectre 3D TOCSY-HSQC
1H–15N permet l’identification des
résidus. Des spins appartiennent au même système lorsqu'ils
présentent entre eux des couplages scalaires. Les protéines ont
cette particularité que les systèmes de spins ne franchissent pas les liaisons peptidiques (pas
de couplage scalaire entre spins de résidus voisins). Tous les protons
couplés avec un
proton amide donné appartiennent donc au même résidu. La
topologie de ce système de spins (nombre et position des
fréquences de résonance des protons) est une
caractéristique qui permet d'identifier la nature des résidus en
présence. Cependant, une dégénérescence certaine
limite à ce niveau l’identification à des familles de résidus.
En pratique, les familles suivantes ont été utilisées :
(Gly), (Ala), (Ser), (Thr), (Val / Ile / Leu), (Glu / Gln / Met / Lys / Arg),
(Cys / Asp / Asn / Phe / His / Trp / Tyr).
Une fois la nature du résidu identifiée pour
chaque pic de l'HSQC 1H–15N, il reste à
déterminer de façon biunivoque sa place dans la séquence.
Cette étape d’attribution est possible grâce au spectre 3D
NOESY‑HSQC
1H–15N. L’effet nucléaire Overhauser
(NOE) diminue avec la distance entre les noyaux considérés. Il se
manifeste particulièrement entre un proton amide et les protons des
résidus voisins. L'exploitation du spectre NOESY‑HSQC 1H–15N
débute par sa comparaison avec le spectre TOCSY-HSQC 1H–15N,
ceci afin de repérer les pics de corrélation internes au
résidu. L'identification des pics restants fournit l'information de voisinage
entre résidus d'acides
aminés. L'identification, même
dégénérée, de la nature des résidus permet
alors de retrouver leur emplacement dans la séquence (Wüthrich, 1986).
Avec cette méthode, la stratégie
d’attribution dépend fortement des éléments de
structure secondaire. L’attribution des hélices
a repose sur les contacts NOE intenses entre protons
amides de résidus voisins dans la
séquence. Les contacts moins intenses entre proton amide et proton a du résidu précédent apporte en outre
une information sur l’ordre des résidus (figure 34).

Figure 34 : Représentation
des contacts NOE au sein de la première hélice a de TolAIII2(0,5 mM). Des bandes horizontales ont
été extraites d’un spectre 3D NOESY–HSQC 1H–15N
à 27°C. Les résonances encadrées correspondent
à celles du spectre 3D TOCSY–HSQC1H–15N.
Les brins b sont eux reconnaissables à l'intensité
des contacts NOE entre proton amide et proton a, ainsi qu'au déblindage significatif de certains
protons a. La topologie des contacts NOE au sein d'un feuillet b est
moins simple que pour une hélices a : bon nombre
de contacts sont observés non entre résidus voisins dans la
séquence, mais entre résidus de brins opposés. En outre, cette topologie est spécifique de la
nature du feuillet b.
Le marquage au 13C de la protéine accélère le travail
d’attribution séquentielle des résonances car il donne
accès à une plus grande variété
d’expériences de RMN. Dans le cas de TolAIII3, j’ai enregistré
un spectre HNCA en plus des spectres 3D TOCSY–HSQC
1H–15N et 3D NOESY–HSQC
1H–15N.
Les résidus
d’acides aminés sont identifiés par leurs
résonances HN et NH (spectre HSQC 1H–15N).
La nature de chaque
résidu est déterminée (avec une certaine
dégénérescence) grâce au spectre 3D TOCSY–HSQC 1H–15N. Le voisinage entre résidus est déterminé grâce au spectre HNCA (Kay et al., 1990).
Un tel spectre corrèle le HN et le NH d’un résidu
avec le Ca propre au
résidu et le Ca du
résidu précédent dans la séquence. Il est donc
possible de proche en proche, de connecter les systèmes de spin voisins
dans la séquence. L’expérience HNCA présente
plusieurs avantages par rapport
à l’expérience 3D NOESY–HSQC
1H–15N. D’abord, un spectre HNCA est facile
d’interprétation puisqu’il comporte seulement deux pics sur
chaque « bande ». Ensuite, comme les corrélations
reposent sur des couplages scalaires
et non pas sur des couplages dipolaires,
elles ne dépendent pas, en première approximation, de la
structure de la molécule dans l’espace (en particulier les
éléments de structure secondaire). Dans le cas où
plusieurs Ca
résonnent à la même fréquence, le recours au spectre
3D NOESY–HSQC 1H–15N
permet en général de lever l’ambiguïté
grâce aux déplacements chimiques des Ha.
Comme précédemment, j’ai extrait une bande
pour chaque N–H peptidique et la série de bandes a
été imprimée pour l’analyse. Par ailleurs,
l’attribution préalable des résonances de la chaîne
principale de TolAIII2 a servi d’appui pour faciliter celle de TolAIII3.
L’attribution des résonances des carbonyles se fait finalement grâce à un spectre
HNCO (Kay et al., 1990)
à partir de l’attribution des résonances N–H.
L’expérience HNCO corrèle le HN et le NH
d’un résidu avec le CO du seul résidu
précédent. Bien que l’on utilise le résidu adjacent
pour l’attribution d’une résonance, cette expérience
ne permet pas d’établir de connectivité entre
résidus.
Un certain nombre d’expériences permettent de
repérer les déplacements chimiques des carbones ou des protons de
chaîne latérale à
partir des résonances NH et HN du même
résidu ou du résidu suivant dans la séquence.
|
Expérience
|
Résonances
observées
sur la chaîne latérale
|
Résonances
NH et HN corrélées
|
|
TOCSY-HSQC
|
protons
|
du
même résidu
|
|
H(CCCO)NH-TOCSY
|
protons
|
du
résidu suivant
|
|
(H)C(CCO)NH-TOCSY
|
carbones
|
du
résidu suivant
|
|
CBCA(CO)NH,
|
Ca et Cß
|
du
résidu suivant
|
|
HNCA
|
Ca
|
du
même résidu et du résidu suivant
|
Tableau 9 : Spectres
tridimensionnels utilisés pour l’identification des
résonances des chaînes latérales
A l’intérieur des systèmes de spin ainsi
identifiés, l’attribution des résonances des carbones et des protons qui leur sont
liés covalemment repose sur l’expérience 3D HC(C)H-TOCSY.
Cette expérience cruciale corrèle un carbone aliphatique, le ou
les proton(s) qui lui sont liés covalemment, et les autres protons du
même système de spin. Par ailleurs, l’attribution
s’appuie en grande partie sur les gammes de déplacements chimiques
propres aux différents carbones et protons de la chaîne
latérale, même si ces gammes peuvent se chevaucher largement (figure 35).
Cette étape de l’attribution s’effectue en
visualisant sur un même écran d’ordinateur les coupes 2D de
tous les spectres 3D ci-dessus, et en vérifiant les alignements à
l’aide d’un curseur lié à tous les spectres. Le
logiciel utilisé ici est FELIX (Accelrys) sur station Silicon Graphics.
A ce stade, l’attribution des résonances
n’est pas stéréospécifique car rien ne permet de distinguer les substituants
de même nature portés par un même carbone.

Figure 35 : Déplacements
chimiques des 1H et 13C des chaînes
latérales.
Diagrammes extraits de (Cavanagh et al., 1996),
en tenant compte des errata publiés sur le site-web du troisième
auteur.
La formation d’un complexe entre deux molécules A et B peut être décrite par
l’équation-bilan suivante et caractérisée par une
constante d’association Ka.
A +
B  AB
AB
Lorsque deux protéines entrent en liaison, la biologie
structurale se donne pour objectif de décrire cette interaction au
niveau moléculaire. Quelles sont les régions des protéines
qui entrent en contact ? Les protéines sont-elles
déformées par l’interaction ? Leur dynamique est-elle
affectée ? Quels sont les paramètres thermodynamiques et
cinétiques de l’interaction ?
La RMN liquide est bien
adaptée à ce type
d’études. En effet, les molécules
d’intérêt se trouve en solution et il est relativement
aisé d’effectuer un titrage de l’association. Il suffit pour
cela d’ajouter des quantités croissantes d’une solution concentrée
de B à une solution de A contenue dans le tube de RMN. Dans
d’autres applications non abordées dans ce travail, la RMN permet
également de cribler simultanément une banque de composés
Bi pour tester si l’un d’entre eux interagit avec la
molécule cible A.
Une des limitations de la RMN étant l’encombrement
des spectres, on évite généralement d’observer
simultanément les protéines A et B. On préfère
observer une seule protéine à la fois. Par exemple, on observera
A et l’effet de B sur A. Il est alors indispensable lors de
l’enregistrement des spectres de pouvoir distinguer
les signaux provenant de A et les signaux
provenant de B. Ceci est rendu possible par un marquage différentiel des partenaires.
L’équation précédente s’écrira alors
(marquage de A) :
A* +
B A*B
Dans notre étude, le
marquage différentiel est obtenu par enrichissement isotopique en 15N
de la seule protéine A*, la protéine B restant froide.
L’expérience HSQC 1H–15N
servant de base à l’analyse des interactions, les signaux
enregistrés proviennent de la seule protéine A*.
VII.C.2. Distinction
des espèces libre et liée
Cependant, la protéine A* peut
être présente sous deux espèces : libre (A*)
ou liée (A*B). Encore une fois, afin de simplifier
l’analyse des spectres de RMN, on privilégiera des conditions
d’études dans lesquelles soit A* soit A*B
est majoritaire
La thermodynamique régit
l’équilibre entre les différentes espèces en
solution.
Soit x la concentration du complexe A*B. Notons x
= [A*B].
Soit a la concentration totale de la protéine
marquée A*. Nous avons a = [A*] + x
Soit b la concentration totale de la protéine B. Nous
avons b = [B] + x
L’équilibre thermodynamique s’écrit alors
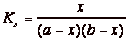 ou encore
ou encore 
Compte tenu de la faible sensibilité de la RMN, l’ordre de
grandeur de a est d’environ 10–3 M. Dans
notre cas, la constante d’affinité Ka est forte,
de l’ordre de 106 M–1. Par
conséquent Ka-1 est
négligeable
devant (a + b). L’équation précédente se
ramène alors à celle–ci : 
qui se résout
aisément pour  ;
;
on obtient x = min(a,b).
Autrement dit, la réaction d’association
est quasi-quantitative. Lors d’un titrage de A* par B la
concentration a est fixe
et la concentration b augmente. Au départ, en l’absence de B,
nous avons [A*B]=0 et [A*] = a. Au cours du
titrage, nous avons [A*B] = b et [A*] = a
– b tant que b reste inférieur à a. Puis [A*B] = a et [A*] = 0. Nous sommes
alors dans des conditions adaptées à l’étude du
complexe A*B.
Pour résumer, lors du titrage suivi par RMN
d’une protéine A* par un ligand B, dans le cas
où la constante d’affinité est très
supérieure à la concentration des protéines, le spectre
initial (avant ajout de B) est caractéristique de la protéine A*
isolée et le spectre final (dès que la quantité totale de
B atteint la quantité initiale de A*), est
caractéristique de la protéine A* liée à
B.
La cinétique
d’échange entre les espèces libre et liée se
reflète dans l’aspect des spectres intermédiaires.
VII.C.3. Phénoménologie
cinétique
La cinétique d’échange peut
être décrite par deux constantes de vitesse : une constante
d’association ka et une constante de dissociation kd.
A* + B
 A*B
A*B
L’évolution des concentrations s’écrit alors

A l’équilibre, ces dérivées sont nulles. On a
donc a kd[A*B] = a ka[A*][B]. La
quantité  peut encore s’écrire kd+ka[B] et
définit la constante d’échange notée kéch.
peut encore s’écrire kd+ka[B] et
définit la constante d’échange notée kéch.
Soit un spin donné de la protéine A*.
Ce spin est en échange entre l’environnement de la protéine
A* libre, dans lequel sa fréquence de résonance est ΩA*, et
l’environnement de la protéine A* liée à
B, dans lequel sa fréquence de résonance est ΩA*B. La vitesse
d’échange entre ces deux environnements est déterminante
pour le type de signal de précession libre observé. Une
description théorique complète des phénomènes
d’échange chimique peut être trouvée dans (Palmer et al., 2001)
En situation d’échange
lent, lorsque kéch
< | ΩA* – ΩA*B |, le signal
de précession libre enregistré pour ce spin est la superposition
des signaux dans les deux environnements. On observe deux pics aux
fréquences de résonance ΩA*
et ΩA*B. L’intensité des pics est proportionnelle
aux concentrations respectives de A* et de A*B (figure
36f). Lors du titrage suivi par RMN de la protéine A* par le
ligand B, on voit apparaître les résonances de A*B
tandis que celles de A* disparaissent progressivement.
En situation d’échange
rapide, lorsque kéch
> | ΩA* – ΩA*B |, le signal
de précession libre enregistré pour ce spin constitue un pic
unique dont la fréquence de résonance est la moyenne des
fréquences de résonance ΩA* et ΩA*B
pondérée par les concentrations respectives de A* et de A*B
(figure 36a). Lors du titrage suivi par RMN de la protéine A*
par le ligand B, on voit les résonances de A* se
déplacer progressivement vers celles de A*B. Dans ces
conditions, il est possible d’attribuer les résonances de A*B
à partir de l’attribution des résonances de A*,
en suivant le déplacement des pics.
En situation d’échange
intermédiaire,
lorsque kéch ≈ | ΩA* –
ΩA*B |, le signal de
précession libre enregistré pour ce spin devient coalescent. Les pics sont larges à tel point
qu’on les distingue faiblement du bruit de fond
(figure 36c). En pratique, on observe donc une disparition des pics de
résonance des spins en échange intermédiaire. Ces
conditions rendent la poursuite de l’étude particulièrement
difficile.

Figure 36 : Spectres
de RMN 1D pour un spin en échange chimique entre deux environnements. De
gauche à droite, les concentrations des composés A* et
A*B sont dans des rapports 3:1, 3:3 et 1:3. De haut en bas, les
constantes d’échanges kéch sont de 10000 s–1
(a), 2000 s–1 (b), 900 s–1 (c), 200 s–1
(d), 20 s–1 (e), 0 s–1 (f).
Les simulations ont été calculées avec des vitesses de
relaxation de 10 s–1.
Figure adaptée de (Palmer et al., 2001)
Plusieurs approches pour automatiser le processus laborieux
d’attribution des contacts NOE ont
été proposées. Dans chacune d’elle, une liste
de candidats à l’attribution
d’un pic de corrélation ambigu est générée
à partir de la position du pic de corrélation, de la liste des
déplacements chimiques des protons de la protéine (la plus
complète possible) et d’un paramètre définissant la
largeur de la plage de déplacements chimiques admis. Ce
paramètre permet de prendre en compte l’incertitude sur le
déplacement chimique, due à la résolution limitée
des spectres, à la forme imparfaite des pics et aux différences
de conditions expérimentales (température, tampon) entre les
enregistrements. Selon la qualité des données, il est souhaitable
de réduire autant que possible cette tolérance car une large plage
de déplacements chimiques admis augmente le niveau
d’ambiguïté.
Le logiciel AutoStructure
((Greenfield et al., 2001)) adopte une approche de
système expert, en implémentant les règles
d’attribution utilisées par un spectroscopiste
expérimenté. Les premières contraintes sont
déterminées sur la base des éléments de structure
secondaire (identifiés grâce aux déplacements chimiques),
sur des considérations de symétrie
dans l’expérience 3D NOESY–HSQC, et sur la consultation de
la carte des contacts attribués. Ce jeu initial de contraintes permet de
calculer un premier modèle moléculaire à partir duquel
d’autres contraintes peuvent être ajoutées de manière
itérative. Les distances entre protons dans le modèle moléculaire
permettent de lever certaines ambiguïtés. Les contraintes violées
sont identifiées et réattribuées.
Dans l’approche de NOAH ((Mumenthaler et Braun, 1995), (Mumenthaler et al., 1997)), les candidats pour un
même pic de corrélation sont traités comme autant de
contraintes de distance indépendantes dans le calcul de structures. Les
contraintes violées dans un grand nombre de structures sont ensuite
éliminées pour le calcul de structures suivant. Le cycle est
répété jusqu’à attribuer chaque pic de
corrélation à une paire unique de protons. Le jeu initial de
contraintes est limité aux pics de corrélations les moins
ambigus.
Dans l’approche d’ARIA
((Nilges et al., 1997),
(Linge et al., 2001)),
les candidats pour un même pic de corrélation sont traités
comme une seule contrainte de distance (ambiguë) dans
le calcul de structures. ARIA considère la contribution de chaque
candidat au pic de corrélation, sans hypothèse a priori. Les candidats dont le pourcentage de contribution
à l’intensité du pic de corrélation,
d’après le modèle structural, est au-dessous d’un
certain seuil sont ensuite écartés à chaque cycle, ce qui
rend la contrainte de distance de plus en plus sélective. Les
contraintes pour lesquelles le nombre de candidats dépassent un certain
plafond d’ambiguïté ainsi que les contraintes violées
systématiquement au-delà d’un certain seuil ne sont pas
prises en compte. Le jeu initial de contraintes comprend la totalité des
candidats (dans la limite du plafond d’ambiguïté maxn).
D’autres approches fortement inspirées d’ARIA
ont également été proposées, qui reposent elles
aussi sur l’utilisation de contraintes de distances
ambiguës.
L’approche de SANE
((Duggan et al., 2001))
utilise une série de filtres dans la sélection des candidats
à l’attribution d’un pic de corrélation ambigu. Outre
leur contribution à l’intensité du pic de
corrélation (critère relatif), les candidats peuvent aussi
être écartés sur la base d’une distance entre protons
trop élevée (critère absolu) dans le modèle
précédent. Par ailleurs, certains candidats peuvent être
éliminés en considérant les éléments de
structure secondaire définis par l’utilisateur. Enfin,
l’éventuelle attribution manuelle d’un pic de
corrélation est considérée et permet
d’éliminer les autres candidats ou d’alerter
l’utilisateur si elle ne figure pas parmi les candidats. Dans cette
approche, une large place est laissée à l’utilisateur pour
contrôler le processus d’attribution.
L’approche du DIEP
(Gilquin et al., 1999), (Savarin et al., 2001) utilise un plafond
d’ambiguïté et un seuil absolu de distance.
L’élimination des autres candidats se fait sur la base de
l’intensité relative de leur contribution au pic de
corrélation. Contrairement à l’usage d’ARIA, la
tolérance sur le déplacement chimique peut être
définie pour chacune des itérations. On peut ainsi
sélectionner lors des premières itérations les pics les
plus faciles à attribuer (tolérance sur le déplacement
chimique et seuil de distance faibles), puis élargir la portée
des attributions possibles (tolérance sur le déplacement chimique
et seuil de distance plus élevés).
Le logiciel CANDID
(Herrmann et al., 2002) reprend le concept de contrainte de distance ambiguë en utilisant une approche
de système expert. Plusieurs règles participent au calcul
d’un score permettant d’éliminer certains candidats. Ces
critères sont : l’écart de déplacements
chimiques entre le pic de corrélation et le déplacement chimique
du candidat, la présence de pics symétriques, la
compatibilité avec la structure covalente (pour les contraintes à
courte portée), l’existence d’un réseau de
contraintes associées et enfin la compatibilité avec les
structures calculées précédemment. Afin de minimiser
l’impact d’une erreur d’attribution, les contraintes à
longue portées sont combinées (sans corrélation
particulière), une contrainte combinée étant satisfaite si
au moins l’une des contraintes individuelles est satisfaite.
Comme il est mentionné plus haut, le fonctionnement
d’ARIA (Nilges et O'Donoghue, 1998),(Linge et al., 2001)
repose sur la notion de contrainte de distance ambiguë. Cette approche considère tous les
candidats pouvant contribuer à l’intensité du pic de
corrélation.
Au cours du calcul de dynamique moléculaire, la contribution
de chaque candidat est
évaluée d’après les distances dans le modèle moléculaire
déjà calculé. La somme de ces contributions est
comparée au volume du pic de corrélation expérimental.
Une telle contrainte ambiguë est satisfaite si au moins un
des candidats est compatible selon le modèle. Cette approche permet
d’intégrer sans biais tous les candidats, sans que ceux qui ne
contribuent pas au pic de corrélation soient connus a priori.
Comme l’information structurale contenue dans une contrainte ambiguë est plus
faible que celle contenue dans une contrainte non-ambiguë, les candidats
ayant les contributions les plus petites sont progressivement identifiés
et éliminés au cours des itérations successives.
Néanmoins, la combinaison de contraintes, même ambiguës,
suffit à orienter le calcul vers la bonne structure, comme
l’illustre l’analogie suivante tirée de (Linge et al., 2001).
Si l’on sait qu’ARIA a été développé
dans un pays germanophone, cette contrainte est ambiguë car il peut
s’agir de l’Allemagne, de l’Autriche ou de la Suisse. Si
l’on sait par ailleurs qu’ARIA a été
développé dans un pays qui borde la mer, cette contrainte est
elle aussi très ambiguë car beaucoup de pays le sont. Pourtant, la
combinaison de ces deux informations ambiguës réduit les
possibilités à un seul pays (l’Allemagne) qui satisfait
à la fois les deux contraintes ambiguës.
ARIA traite indifféremment des pics de
corrélation NOE explicites
(attribués manuellement) ou des pics de corrélation NOE implicites (définis seulement par les
déplacements chimiques de protons et de l’éventuel
hétéroatome). L’implémentation
locale d’ARIA a été modifiée (fichier NoeList.py) afin de traiter
également les pics partiellement attribués
dans les expériences 3D NOESY-HSQC.
Chaque itération
utilise la même liste de pics de corrélation. Seules les
structures (de plus basse énergie) sont héritées
d’une itération sur la suivante et affectent les étapes de
calibrage et d’attribution automatique des pics.
Au début de chaque itération, chaque spectre est
calibré indépendamment. Un spectre NOE est
rétro-calculé à partir des meilleures structures de
l’itération précédente et comparé au spectre
expérimental afin de déterminer des constantes de calibrage (par
types d’atomes). Les volumes expérimentaux des pics de corrélation
sont alors traduits en contraintes de distance (éventuellement
ambiguës). Les meilleures structures de l’itération
précédente sont à nouveau analysées pour
déterminer les contraintes violées systématiquement. Cette
étape de calibrage est effectuée une première fois avec
toutes les contraintes puis une seconde fois sans les contraintes
violées. Enfin, les
bornes des contraintes violées sont élargies à
l’intervalle [0 ; 6] Å et les contraintes toujours
violées sont éliminées. Les violations sont définies
par deux seuils de tolérance (en distance et en proportion sur
l’ensemble structural) qui peuvent être paramétrés
par l’utilisateur (paramètres violtoler et violratio).
L’étape suivante est l’attribution
partielle des contraintes ambiguës.
Les contraintes acceptées à ce stade sont analysées au
regard des meilleures structures de l’itération
précédente afin de déterminer la contribution de chaque
paire de protons à l’intensité du pic de
corrélation. Seules les contributions majoritaires sont conservées.
Le paramètre ambigcutoff détermine la proportion des
contributions conservées.
Après les étapes de
calibrage et d’attribution, les contraintes des différents spectres NOESY sont écrites dans le
répertoire de l’itération puis rassemblées (en éliminant les redondances) avant
d’être intégrées aux autres contraintes (angles
dièdres, liaisons hydrogène...) dans un calcul de
dynamique moléculaire (recuit
simulé) à partir des meilleures structures de
l’itération précédente et de structures
aléatoires. Les nouvelles structures calculées serviront de base
à l’itération suivante.
La liste de
diffusion aria-discuss
rassemble les remarques des utilisateurs d’ARIA.
ARIA est constitué d’une série de scripts
Python et utilise CNSsolve (Brunger et al., 1998). Les fichiers
nécessaires au bon fonctionnement d’ARIA version 1.0 ont
été installés dans le répertoire /lx1/people/deprez/aria1.0/
.
ARIA requiert l’installation de CNSsolve (incluant les
fonctions ARIA) et d’un interpréteur Python.
CNSsolve version 1.0 (http://cns.csb.yale.edu/v1.0/)
a été compilé pour les stations SGI (répertoire /ric1/people/deprez/acns_solve_1.0/) et pour les stations Linux
(répertoire /lx1/people/deprez/acns_solve_1.0/) en incluant les fonctions ARIA. Pour le
bon fonctionnement de CNSsolve, les lignes suivantes doivent être
incluses dans le fichier .cshrc de
l’utilisateur :
if (( $HOST == 'rmnlx1' ) || (
$HOST == 'rmnlx2' )) then
setenv CNS_SOLVE '/lx1/people/deprez/acns_solve_1.0/'
else
setenv CNS_SOLVE '/ric1/people/deprez/acns_solve_1.0/'
endif
alias acns
'source $CNS_SOLVE/cns_solve_env; nice +15 cns_solve'
Des problèmes de calibrage de l’itération 0
sont rencontrés avec la version de CNSsolve compilée pour Linux
(messages 99 et suivants de aria-discuss).
Un interpréteur
Python (http://www.python.org)
incluant le module Numeric a été installé
dans le répertoire /usr/local/bin/
. Les lignes suivantes doivent être incluses dans le fichier .cshrc de
l’utilisateur :
alias python
'/usr/local/bin/python1.6'
setenv
PYTHONPATH /lx1/people/deprez/aria1.0/
ARIA est lancé par la commande :
/usr/local/bin/python
/lx1/people/deprez/aria1.0/Aria/RunAria.py
Si l’on
veut pouvoir se délogger sans interrompre ARIA, on utilisera la
commande :
nohup /usr/local/bin/python
/lx1/people/deprez/aria1.0/Aria/RunAria.py >> ./run.out &
Il est utile de créer des raccourcis pour ces commandes
dans le fichier .cshrc
de l’utilisateur.
Les scripts CGI
pour l’utilisation d’une interface graphique HTML ont été installés sur le
serveur hamlet
dans le répertoire /var/www/cgi-bin/aria/
. La page d’accueil est accessible à l’adresse
suivante :
/lx1/people/deprez/aria1.0/html/aria.html
.
J’ai créé
dans le répertoire /lx1/people/deprez/aria1.0/perlscripts/
une série de scripts
additionnels pour la préparation et l’analyse des données (Noehsqc_ppm_maker_n15.pl, Noehsqc_ppm_maker_c13.pl, Flx2List.pl,
Bmrb2Talos.pl, Bmrb2seq.pl, Bmrb2ppm.pl, ADRanalysis.pl, ICount.pl,
Tbl2Sheet.pl, PdbMmAlign2.pl, PdbRmsd.pl, DiscoAriaAlign.pl, energies.csh).
L’utilisation de ces scripts est décrite plus loin. En
l’absence de précisions supplémentaires, la lecture des
données pour ces scripts se fait sur l’entrée standard et
l’écriture des données se fait sur la sortie standard. Il
est possible de rediriger ces deux flots grâce aux opérateurs UNIX
‘ < ‘ et
‘ > ‘
respectivement.
i) Contraintes de
distances
Les spectres NOESY ont été analysés avec le
logiciel FELIX (Accelrys Inc.).
Il est possible de préparer automatiquement une liste de pics de corrélation intrarésiduels et/ou séquentiels au
format FELIX à partir de la table de déplacements
chimiques au format STAR (BMRB) en
utilisant le script PERL Noehsqc_ppm_maker_n15.pl
pour l’expérience 3D NOESY–HSQC 1H–15N
(corrélations intrarésiduelles et séquentielles) et le
script PERL Noehsqc_ppm_maker_c13.pl
pour l’expérience 3D NOESY–HSQC 1H–13C
(corrélations intrarésiduelles).
Les listes de
pics 3D au format FELIX peuvent ensuite
être converties au format
LIST utilisé par ARIA
grâce au script PERL Flx2List.pl. On aura pris soin, pour les pics de corrélation attribués manuellement, de
respecter la nomenclature IUPAC (figure 37) et la syntaxe suivante. Les contraintes dont les annotations
incluent le caractère ‘ % ‘ ne sont pas prises en compte.
L’annotation ‘null’
désigne un pic non attribué explicitement. Les
éléments d’une attribution (type de résidu,
numéro de résidu, nom d’atome dans cet ordre) sont
séparés indifféremment par le caractère
‘ _ ‘
(underscore) ou le caractère ‘ : ’. Le type de résidu est
optionnel, ainsi que le caractère H des protons autres que le proton
amide. Plusieurs attributions peuvent être accolées. Il
n’est pas nécessaire de répéter le numéro de
résidu lorsque plusieurs atomes du même résidu participent
à une attribution. Le caractère ‘ * ‘ peut
être utilisé pour remplacer n’importe quel caractère.
Ainsi, la nomenclature HD*
désigne indifféremment les deux protons HD2 et HD3. Il est rappelé que
l’implémentation de FELIX n’autorise pas plus de 16
caractères pour désigner une attribution. On réservera
donc cette méthode aux pics de corrélations présentant un faible
nombre de candidats. Voici quelques exemples de syntaxe correcte :
|
%
|
ALA_77_HB
|
LYS_78_HD*
|
90_HA_87_HA
|
16_D1_46_D1_G2
|
|
null
|
LYS_78_HB3
|
63_HB3
|
88_HE*_HD*_90_H
|
26_B_30_D3_92_D2
|

Figure 37 : Nomenclature
atomique pour les résidus d’acides aminés,
recommandée par l’Union Internationale de la Chimie Pure et
Appliquée (IUPAC). Figure reproduite de (Markley et al., 1998)
Le script Flx2List.pl
fait appel à un fichier vol.txt contenant les volumes de pics
exportés depuis FELIX et un fichier seq.txt contenant la
séquence peptidique (un résidu par ligne sous forme de trigramme
en majuscules). Ces fichiers doivent être placés dans le
répertoire courant. Le fichier seq.txt peut être créé avec Bmrb2seq.pl à partir
de la table de déplacements chimiques au format STAR (BMRB).
La nomenclature atomique utilisée par le champ de force
(issu de CHARMM) étant différente de celle utilisée dans
la base de données de déplacements chimiques BMRB, une conversion est effectuée par le script Flx2List.pl. La
correspondance entre les deux nomenclatures est indiquée ci-dessous
(uniquement pour les nomenclatures des protons qui diffèrent).
|
Résidu
|
Nomenclature
BMRB
|
Nomenclature
CHARMM
|
|
Tous excepté PRO
|
H
|
HN
|
|
GLY
|
HA2
HA3
|
HA1
HA2
|
|
ASN, ASP, CYS, HIS,
PHE, SER, TRP, TYR
|
HB2
HB3
|
HB1
HB2
|
|
GLN, GLU
|
HB2
HB3
HG2
HG3
|
HB1
HB2
HG1
HG2
|
|
ARG, PRO
|
HB2
HB3
HG2
HG3
HD2
HD3
|
HB1
HB2
HG1
HG2
HD1
HD2
|
|
MET
|
HB2
HB3
HG2
HG3
HE
|
HB1
HB2
HG1
HG2
HE1, HE2, HE3
|
|
LYS
|
HB2
HB3
HG2
HG3
HD2
HD3
HE2
HE3
HZ
|
HB1
HB2
HG1
HG2
HD1
HD2
HE1
HE2
HZ1, HZ2, HZ3
|
|
ALA
|
HB
|
HB1, HB2, HB3
|
|
THR
|
HG2
|
HG21, HG22, HG23
|
|
VAL
|
HG1
HG2
|
HG11, HG12, HG13
HG21, HG22, HG23
|
|
ILE
|
HG12
HG13
HG2
HD1
|
HG11
HG12
HG21, HG22, HG23
HD11, HD12, HD13
|
|
LEU
|
HB2
HB3
HD1
HD2
|
HB1
HB2
HD11, HD12, HD13
HD21, HD22, HD23
|
Figure 38 : Différences
de nomenclature entre la base de données de déplacements
chimiques BMRB et le champ de force utilisé par ARIA pour les protons
des résidus d’acides aminés.
La variable $WHICH_PEAKS
en tête du script Flx2List.pl définit quel type de contraintes seront
converties. La valeur “all“
entraîne la conversion des contraintes explicites
et implicites. La valeur “allassi“ entraîne la conversion
des contraintes explicites seulement.
ii) Contraintes diédrales
Le fichier
d’entrée pour TALOS
(Cornilescu et al., 1999) peut être créé
à partir de la table de déplacements chimiques au format STAR (BMRB) en utilisant le script PERL Bmrb2Talos.pl. Ce script
fait appel au fichier seq.txt placé dans le répertoire
courant. Il effectue le changement de référence des
déplacements chimiques pour passer du DSS au TSP.
Les données
issues de TALOS peuvent
être converties au format .tbl en utilisant le script awk talos_to_xplor.gawk fourni
avec TALOS.
iii) Table de déplacements chimiques
La table de déplacements chimiques utilisée par
ARIA peut être créée
à partir de la table de déplacements chimiques au format STAR (BMRB) en utilisant le script PERL Bmrb2ppm.pl. Ce script
opère la conversion de nomenclature
des atomes évoquée précédemment.
L’utilisation d’ARIA comprend 4 étapes successives résumés sur la figure 39.

Figure 39 : Déroulement
d’ARIA. Les extensions de fichiers correspondent aux types de
données suivantes. seq : séquence peptidique ;
list : contraintes de distances dans le format ARIA ;
ppm : table de déplacements chimiques ; py : script
Python ; tbl : contraintes expérimentales dans le format
CNS ; cns : paramètres pour CNS ;
psf : topologie moléculaire ; pdb : structure
tridimensionnelle ; inp : script pour CNS ;
out : journal de sortie de CNS
La première
étape d’un calcul avec ARIA
consiste à créer un fichier new.html . Ce fichier contient les
informations nécessaires à la préparation des
données (identifiant du calcul,
chemins d’accès des principaux fichiers, largeurs
des plages de déplacements chimiques admis, mode de
pondération des contraintes de distances, etc.). Ce fichier peut
être créé à partir d’un formulaire
HTML accessible à l’adresse suivante :
/lx1/people/deprez/aria1.0/html/aria.html.
A l’usage, l’utilisateur constatera qu’il est plus rapide de
modifier directement un fichier existant plutôt que de recréer un
fichier ab initio.
La deuxième étape consiste à lancer ARIA dans le
répertoire qui contient le fichier new.html . Ceci a pour effet d’établir
l’arborescence pour le run
et de créer une copie locale des données (sous-répertoire data) et des protocoles (sous-répertoire protocols).
L’archivage est ainsi assuré même si les données et
protocoles originaux viennent à être modifiés
ultérieurement. Les listes de contraintes de distances au format .list sont converties au format
.tbl propre à
CNS.
La troisième
étape consiste à
créer dans le répertoire du run un fichier run.cns. Ce fichier inclut tous les paramètres
nécessaires au calcul
itératif : types de contraintes, pont disulfure, spectres
utilisés, matrice de relaxation, paramètres pour les
itérations (maxn, violtoler,
violratio, ambigcutoff...),
protocoles de recuit simulé, etc. Un fichier run.cns par défaut a
été créé lors de la deuxième étape.
Ce fichier peut être édité à partir du formulaire
HTML accessible à l’adresse :
/lx1/people/deprez/aria1.0/html/aria.html.
Comme précédemment, l’utilisateur constatera qu’il
est plus rapide de recopier et retoucher un fichier préexistant
plutôt que de recréer un fichier ab initio.
La quatrième étape consiste à lancer ARIA dans le
répertoire du run
(celui qui contient le fichier run.cns
). Cela enchaîne les protocoles CNS sans autre intervention de
l’utilisateur. Après les 8 itérations, les meilleures structures sont affinées dans une couche d’eau.
i) Analyse de l’attribution
semi-automatique des contraintes de distance
Le script PERL ADRanalysis.pl
permet l’analyse des contraintes de distance lors des étapes de
calibrage et d’attribution
d’une itération. Ce script doit être exécuté
dans le sous-répertoire de l’itération avec comme arguments
le nom du jeu de contraintes à analyser (généralement le
nom du spectre dont il est issu) et le numéro de
l’itération. Il apparaît alors de gauche à droite et
pour chaque contrainte :
·
le statut initial de la contrainte (1=non-ambiguë
acceptée, 2=non-ambiguë violée, 3=ambiguë
acceptée, 4=ambiguë violée, 5=non-utilisée,
6=nouvelle)
·
le numéro de la contrainte dans la liste de pics
·
le niveau d’ambiguïté initial
(‘-‘=implicite, 1=non-ambigu, 2=2 candidats, etc.)
·
‘D’ pour un pic diagonal ou
‘ ? ‘ pour une erreur de sélection (pas de
déplacement chimique dans la plage souhaitée)
·
‘X’ si un problème a
été rencontré avec la contrainte (examiner alors le
journal de sortie du calibrage)
·
‘%’ pour chacune des meilleures structures
de l’itération précédentes pour laquelle la
contrainte est violée (3 passages).
·
‘X’ lorsque la contrainte est exclue
·
le statut final de la contrainte (1=non-ambiguë
acceptée, 2=non-ambiguë violée, 3=ambiguë
acceptée, 4=ambiguë violée, 5=non-utilisée,
6=nouvelle)
·
le numéro de la contrainte dans la liste de pics
·
le niveau d’ambiguïté après
attribution (1=non-ambigu, 2=2 candidats, etc.)
·
‘S...’ lorsque la contrainte est
utilisée pour le calcul de structure.
Ce listage peut ensuite être filtré avec la
commande UNIX grep
pour des besoins statistiques. Le script PERL ICount.pl permet d’afficher à partir de
ce listage l’histogramme des
contraintes en fonction de leur niveau d’ambiguïté
après attribution.
Le script PERL Tbl2Sheet.pl
produit, à partir d’un fichier .tbl d’itération, une feuille de
calcul permettant le décompte et la
classification des contraintes à l’aide d’un tableur. Chaque atome impliqué dans une contrainte
possède une entrée avec les informations suivantes : le
numéro de la contrainte, le caractère non-ambigu (U) ou de
stéréochimie flottante (S) ou ambigu (A) de la contrainte, le nom
du résidu de l’atome, le numéro du résidu de
l’atome, le nom de l’atome, le nom du résidu de
l’atome partenaire, le numéro du résidu de l’atome partenaire,
le nom de l’atome partenaire, la distance dans la séquence entre
les deux résidus. Par symétrie, chaque contrainte apparaît
donc en double et une variable booléenne est également
créée permettant de filtrer les redondances.
ii) Analyse de
l’ensemble structural calculé
Le script energies.csh
affiche le listage des énergies
(totale, de liaison, angulaire, diédrale, de van der Waals, NOE...) de
chacune des structures calculées dans l’itération courante, par ordre
d’énergie totale décroissante. Les noms des fichiers .pdb
correspondants se trouvent dans les fichiers file.nam et file.list .
Le script PERL PdbMmAlign2.pl
crée à partir de la liste de structures contenue dans le fichier file.nam une macro pour le
logiciel MOLMOL (Koradi et al., 1996)
permettant de générer un fichier molmolfit.pdb qui contient un alignement de l’ensemble
structural. Ce fichier est utilisé
par le script PERL PdbRmsd.pl
pour calculer les écarts-types à la moyenne et les écarts-types croisés, calculés sur la position de la chaîne
principale des résidus définis en tête de script.
Le script PERL DiscoAriaAlign.pl
génère une macro pour le logiciel Insight (Accelrys Inc.)
permettant d’afficher un alignement
des structures de meilleure énergie
de l’itération. La variable $ENSEMBLE en tête de script
définit le nombre de structures à aligner. La variable $SUBSEQ
définit la zone d’alignement. La variable $ROOTNAME définit
la racine de nommage des objets.
Les échantillons de scones ont été
préparés selon le mode opératoire suivant. 2,5 g de
chlorure de sodium, 4 g de poudre à lever (tartre 60%; bicarbonate de
sodium 25%; glucose polymérisé 15%) ont été
mélangés à 200 g de poudre de gamètes femelles
fécondés de Triticum aestivum
(débarrassés de leur enveloppe). 50 g de résidu lipidique
(battu en baratte) de sécrétion mammaire de Bos taurus ont été dissous manuellement dans ce
mélange puis 25 g de saccharose ont été additionnés
avant homogénéisation du fluide. 1 ovocyte de Gallus
gallus (débarrassé de son
enveloppe calcique et broyé à l'aide d'une tige métallique
portant 4 pointes parallèles à son extrémité) et
0,2 L de sécrétion mammaire de Bos taurus ont été ajoutés. Le
mélange a été pressé manuellement jusqu'à
obtention d'un volume déformable non-adhérent, lequel a
été adsorbé sur un plan horizontal sur une épaisseur
de 0,015 m. Des disques ont été extraits à l'aide d'un
cylindre évidé de diamètre 0,07 m. Ils ont
été recouverts d'une couche mince uniforme de broyat d'ovocyte de
Gallus gallus (vide supra) et
disposés sur une plaque métallique lubrifiée
(résidu lipidique battu en baratte de sécrétion mammaire
de Bos taurus). Les
échantillons ont été portés à une
température de 473 K pendant une durée suffisante pour permettre
un accroissement de volume d'un facteur 2 à 3 sans modification
significative du spectre d'absorption de la surface ni dégagement de
particules volatiles en quantités excessives, puis ils ont
été laissés à température ambiante.
La version originale de ce
document a été rédigée avec Microsoft Word 98 pour
Macintosh (http://www.microsoft.com/)
dans le respect des styles
décrits dans le « Guide pour la rédaction et la
présentation des thèses à l’usage des
doctorants »édité
par le Ministère de l’Education Nationale et le Ministère
de la Recherche en 2001 (téléchargeable à l’adresse http://www.sup.adc.education.fr/bib/Acti/These/guidoct.rtf).
Les caractères soulignés constituent des liens hypertexte.
Les références
bibliographiques ont été
gérés avec ProCite 4 pour Macintosh (http://www.procite.com/).
Les figures ont été converties dans le format
JPEG avec GraphicConverter pour Macintosh (http://www.lemkesoft.com/).
Les figures 10, 16 et 7, 8, 13b ont
été construites sous UNIX respectivement avec MolScript (Kraulis, 1991) http://www.avatar.se/molscript/ et
Raster 3D (Merritt et Bacon, 1997) http://www.bmsc.washington.edu/raster3d/raster3d.html,
à partir des fichiers de coordonnées déposés dans
la base de données Protein Data Bank (http://www.rcsb.org/pdb/index.html).
La figure de couverture (représentation en saucisses) a
été réalisée avec MOLMOL (Koradi et al., 1996)
http://www.mol.biol.ethz.ch/wuthrich/software/molmol/
à partir de l’ensemble structural calculé dans la partie V.
Les articles joints au document sont dans le format PDF et
peuvent être lus avec le logiciel Adobe Acrobat Reader
(téléchargeable gratuitement sur le site de
l’éditeur http://www.adobe.com/).